
Solving the Data Analysis Bottleneck of Modern Proteomics: A Software Tool That Replaces Three Separate Steps With One Unified Method

A scalable matrix factorization method from Ghent University integrates imputation, batch correction, and dimensionality reduction in proteomics into a single open-source R/Bioconductor package, increasing statistical power and dramatically improving data processing
Executive Summary
Proteomics is the science of measuring the proteins present in biological samples — the actual molecules that perform the functions of life inside cells. Where genomics tells you which genes a cell could express, proteomics tells you which proteins the cell actually produces and in what quantities. Modern proteomics relies on mass spectrometry, an instrumental technique that ionises proteins and measures their masses with extraordinary precision, identifying thousands of distinct proteins in a single sample within minutes. The latest generation of mass spectrometers can profile the proteome of a single human cell — a measurement that was technically impossible a decade ago — and has unlocked applications across pharmaceutical research, clinical diagnostics, biotechnology, agricultural science, and forensic analysis.
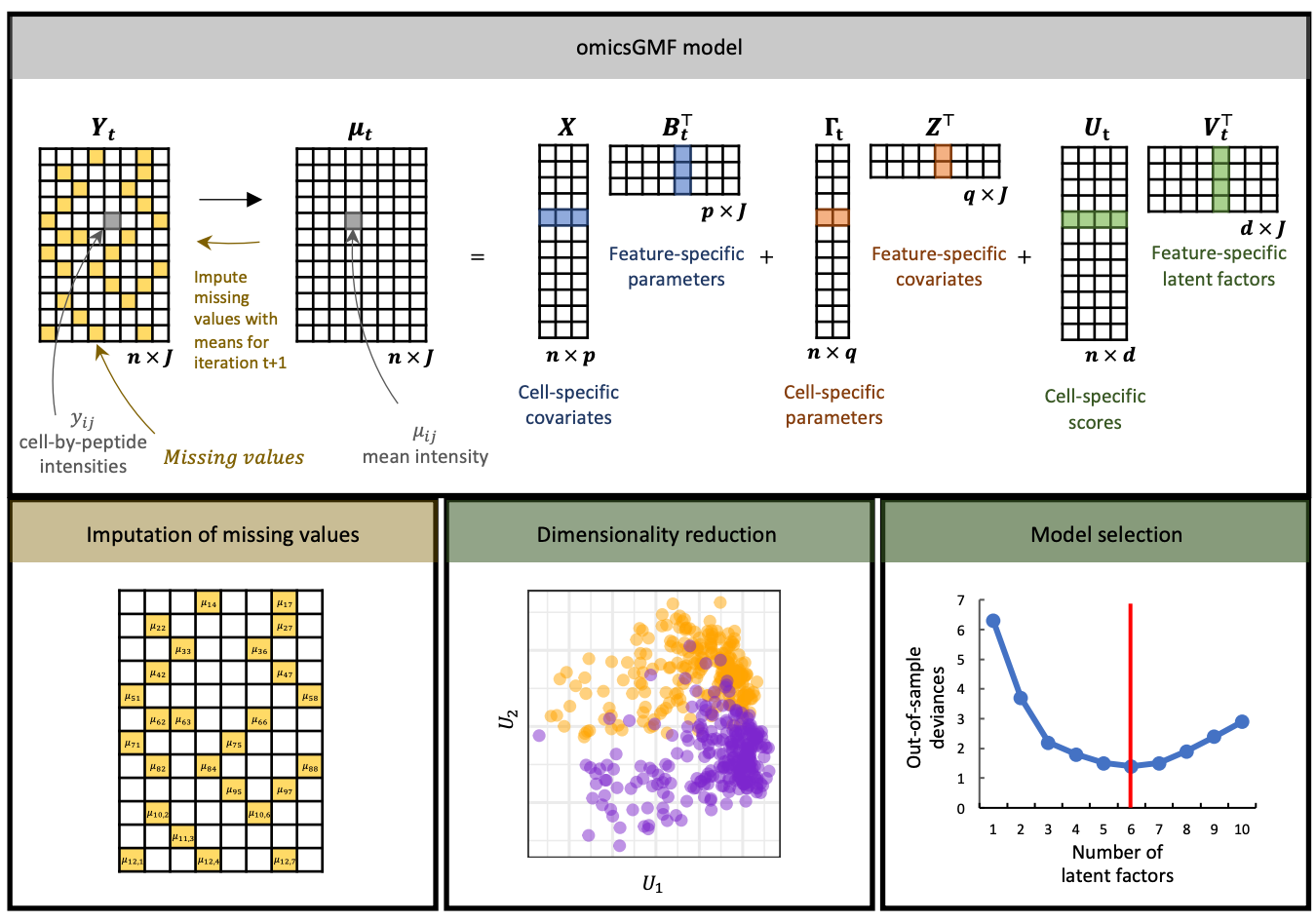
This instrumental revolution has created a corresponding bottleneck in data analysis. A modern proteomics experiment generates massive datasets that suffer from three interrelated problems simultaneously. The first is batch effects: small variations in instrument performance, sample preparation, and reagent batches across multiple measurement runs introduce systematic biases that obscure the real biological signal in the data. The second is missingness: not every protein is detected in every sample, so the resulting data matrices are full of gaps that complicate statistical analysis. The third is dimensionality: a single proteomics dataset can contain measurements of thousands of proteins across hundreds or thousands of samples, generating millions of data points whose interpretation requires sophisticated mathematical reduction to a small number of meaningful summary dimensions.
The current standard workflow addresses these three problems sequentially: first impute the missing values, then correct for batch effects, then apply dimensionality reduction. Each step uses a different software tool, each tool has its own assumptions and parameters, and the sequential application creates compounding errors. The data is processed three times, and small mistakes at each step accumulate into larger distortions in the final analysis. The research published in Nature Communications on 20 May 2026 by Segers, Castiglione, Clement and colleagues at Ghent University addresses this bottleneck directly. The authors present omicsGMF, an R/Bioconductor software package that integrates all three operations — imputation, batch correction, and dimensionality reduction — into a single unified mathematical framework based on matrix factorization. The integrated approach delivers two quantitative outcomes. First, it produces more accurate imputation of missing values than the best existing dedicated imputation tools. Second, it increases the statistical power of downstream analyses to detect differentially abundant proteins — the proteins whose levels actually differ between experimental conditions, which are the biological discoveries that proteomics experiments are designed to find. For pharmaceutical companies, clinical diagnostics providers, biotechnology firms, and instrumentation vendors, this is an open-source tool that immediately improves the value extracted from every proteomics experiment.